蛋白質組學概念的起源和發展
蛋白質組學的誕生和發展,離不開多學科和技術的逐漸交叉融合。這些學科技術包括(但不限于)基因組學、生物化學、分析化學、自動化、基于電磁場的精密質譜儀、信號處理、數理統計和計算機科學。近年來,分子醫學、大數據技術和人工智能的發展,進一步加速推動了蛋白質組學的成長,使之在精準醫療領域展示出越來越大的應用潛力。

01蛋白質組學的華麗誕生
1994年,當27歲的博士研究生馬克·威爾金斯(Marc Wilkins)在地廣人稀的澳大利亞嘗試把蛋白質(protein)和基因組(genome)拼成一個新的英語單詞蛋白質組(proteome),用以描述基因組編碼的所有蛋白質,并將這一單詞放在他的博士畢業論文中時,他不會想到,3年后,瑞士蘇黎世聯邦理工大學的皮特·詹姆斯(Peter James)在他發表于劍橋大學《生物物理學》季刊的一篇53頁的長文中借用了這個概念,并首次提出蛋白質組學(proteomics)一詞,系統總結了當時已發表的對生物體內所有蛋白質種類的研究以及該類研究的進展。

蛋白質組學不是憑空誕生的一個新學科,而是基于一系列蛋白質的生物化學研究和多肽質譜的研究衍生發展而來的,所有這些相關學科的研究都被串起來,成為這一新學科的基石。
威爾金斯更不會想到蛋白質組學會受到如此廣泛的關注。1997年,人類基因組計劃進行得如火如荼,2001年,《科學》(Science)雜志和《自然》(Nature)雜志分別出版專刊,報道了人類基因組計劃草圖的完成,興奮地宣告解讀了人類生命的編碼。
生命科學的中心法則清楚地表明,基因只是遺傳編碼,在生命活動中真正發揮作用的主要是蛋白質。
因此,在《科學》雜志報道人類基因組計劃完成的專刊上,華盛頓大學的斯坦利·菲爾茨(Stanley Fields)預言蛋白質組學將很快取代基因組學成為生命科學研究的焦點;
《自然》雜志的專刊則在顯著版面報道了人類蛋白質組學組織(HUPO)的成立,并宣告生命科學正式進入蛋白質組學時代。因為人類基因組計劃的巨大成功,蛋白質組學在誕生之初,光環熠熠,世界各國對蛋白質組學予以大量投入,工業界也熱情洋溢,不可謂不華麗。
現在,回顧蛋白質組學的華麗誕生,我們感情復雜。一方面,基因組學的巨大成功讓全世界認識到蛋白質組學毋庸置疑的重要性;另一方面,蛋白質組學直到今天也還沒有完全擺脫基因組學的巨大成功映射出的陰影帶來的困擾。例如,在相當長一段時間內(直到今天仍然存在),蛋白質組學的別名是“功能基因組學”,因而其常常被列為基因組學的一部分而存在。
02基因組學巨大成功背后的陰影
人類基因組計劃是將基因按照染色體的分布承包給各個研究團隊,協同開發技術,分別測序,然后拼裝成全基因組。這個化整為零、逐個擊破的簡單思路取得的成功,是迄今為止全世界不同國家的科學家互相協作、進行超大項目研究的成功典范。
早期參與人類蛋白質組計劃的研究人員蕭規曹隨,選擇了同樣的思路,將蛋白質組按照染色體分組,然后分配給世界各國的參與團隊。
后來的數據表明,這個復制和迎合基因組學的思路在蛋白質組學領域并未獲得同行的一致認可,也沒有取得其他生命科學領域的認可。
基于染色體的蛋白質組研究確實取得了不少成果,但與巨大的資金和熱情投入而急速鼓吹起來的期望值相比,這些成果微小得幾乎不能被人們看到。這一段至今尚未完全結束的歷程,極大地消費了人們對蛋白質組學的期望和熱情。
蛋白質組學的華麗誕生在其第一個10年感受到了全球各界的熱情,出現了一段時間繁榮的景象:學術界和工業界的大量投入,專業雜志接二連三地涌現,影響因子逐年升高。今天,當我們擁有了高精度質譜儀和比較完善的算法后,回顧歷史,我們不得不汗顏地承認:當時很多蛋白質組學研究所產生的數據信息量是非常低的,有些甚至是經不起時間考驗的。
2014年,《自然》雜志倉促地發表了兩篇號稱是完成了人類蛋白質組草圖的論文,認為其代表了當時蛋白質組學的最好研究。這兩項研究在若干種不同人緣樣本中對超過17000個蛋白質進行了鑒定,給觀望蛋白質組學的大眾打了兩針興奮劑。
但是后來,蛋白質組研究領域的多位同行對這兩篇論文中所使用的數據分析方法提出了質疑,并證明其中有些數據是錯誤的,從而引發了大量的后續討論。事實上,僅僅在多種樣本中鑒定到這些蛋白質的表達,而不對它們進行精確的定量,并不會產生太大的生物學價值。
換言之,僅僅讓大家看到蛋白質組學在經過17年的努力后終于在蛋白質鑒定水平達到了基因組測序覆蓋率的70%(暫不考慮多肽水平的覆蓋率),勉強及格,只是進一步加深了大家對“蛋白質組學從屬于基因組學”這一誤區的認同而已。
03蛋白質組學的牛刀小試
鑒于蛋白質的復雜性和多變性,完全意義上的蛋白質組學至今仍是一個科學目標或者科學理想,因為至今無人知道一個生物體內到底有多少蛋白質。比蛋白質組學本身更加繁榮的,并令所有人毫無爭議的、振奮的乃是色譜-質譜方法學的巨大進展。
色譜-質譜技術在過去的20年高速發展,越來越多的生物醫學科學家的研究受益于色譜-質譜技術的發展,比如未知蛋白質的鑒定、蛋白質相互作用的鑒定、翻譯后修飾的鑒定、蛋白質結構的解析、靶向蛋白質定量、蛋白質降解的研究等。有些方法比如靶向蛋白質組學,正在走向臨床試驗。但是嚴格意義上講,這些都不是蛋白質組學的主要內涵。令人尷尬的是,雖然色譜-質譜技術取得了長足的進步,但蛋白質組學這一學科卻逐漸淡出主流研究的視野。
蛋白質組學不是沒有獲得過大眾認可的成功。比如,基于同位素標記的定量蛋白質組學可以對2~4個樣本的蛋白質組進行準確定量,在進行良好的實驗設計和實施后,8000個以上的蛋白質(基因產物)可以被鑒定到,并且含有準確的定量信息,進而引導新的生物學發現。雖然這些成功往往只出現在一部分擁有高超實驗技巧的蛋白質組學實驗室,但這已經可以讓大眾慢慢意識到蛋白質組學在生物研究中實實在在的強大力量,從而獲得了一部分支持。實際上,跟蛋白質組學博大的內涵相比,這些成功只能算是牛刀小試。
04蛋白質組學和精準醫療
人類的幾乎所有生命活動都是由人體內的蛋白質執行的。人類的健康和疾病同蛋白質息息相關,而疾病治療的效果也取決于蛋白質機器的調控。所有熟悉生物學中心法則的大眾應該沒有人會質疑蛋白質在精準醫療中不可替代的作用。
蛋白質組學作為研究所有蛋白質的科學,毫無疑問將在精準醫療領域發揮最關鍵的作用。然而,直到最近,這些作用還只能被稱為“潛力”。
蛋白質組學發展到今天,才剛剛走過21個年頭。被撇在基因組巨大的身形背后,21歲的蛋白質組學常常有意無意被人遺忘,或者被認為是可有可無的“跟班”或“錦上之花”。生物學的中心法則在基因組的燦爛光環下黯然失色。
基因組學在種類眾多但數量有限的遺傳性單基因疾病和產前診斷中展示了毫無爭議的作用后,一般被大眾誤解為精準醫療的主要甚至是唯一的方式。
筆者認為,對基因組學與其實際生物學功能不相稱的期望,為今后基因組學在數量更多的人類復雜疾病(比如絕大部分腫瘤、代謝性疾病、心腦系統疾病等)中的臨床應用的跌宕,埋下了伏筆。
近年來,越來越多的科學家開始重新思考蛋白質組學在精準醫療中的應用,并且一系列切實的蛋白質組項目正在開展。
雖然年輕的蛋白質組學已經經歷了一系列盛衰榮辱,但其成長在跌宕起伏中一刻也未停止過,尤其是近5年來,已在各個技術環節取得了突破性的進展。
現在,我們已經有新技術可以對極小量的臨床樣本進行高通量的、快速、準確的蛋白質組學水平的定量,并且在越來越多的臨床應用中展示出獨特的、有效的作用,主流生命科學界和醫學界的關注與日俱增,其他領域比如醫療大數據和人工智能的研究人員也展示出了極大的興趣。
蛋白質組(Proteome)的概念最先由Marc Wilkins提出,指由一個基因組(Genome),或一個細胞、組織表達的所有蛋白質(protein). 蛋白質組的概念與基因組的概念有許多差別,它隨著組織、甚至環境狀態的不同而改變。 在轉錄時,一個基因可以多種mRNA形式剪接,一個蛋白質組不是一個基因組的直接產物,蛋白質組中蛋白質的數目有時可以超過基因組的數目。 蛋白質組學(Proteomics)處于早期“發育”狀態,這個領域的專家否認它是單純的方法學,就像基因組學一樣,不是一個封閉的、概念化的穩定的知識體系,而是一個領域。
主要功能
蛋白質組學集中于動態描述基因調節,對基因表達的蛋白質水平進行定量的測定,鑒定疾病、藥物對生命過程的影響,以及解釋基因表達調控的機制. 作為一門科學,蛋白質組研究并非從零開始,它是已有20多年歷史的蛋白質(多肽)譜和基因產物圖譜技術的一種延伸. 多肽圖譜依靠雙向電泳(Two-dimensional gel electrophoresis, 2-DE)和進一步的圖象分析;而基因產物圖譜依靠多種分離后的分析,如質譜技術、氨基酸組分分析等.
由于可變剪輯及RNA編輯的存在,許多基因可以表達出多種不同的蛋白質。因此,蛋白質組的復雜度要比基因組的復雜度高得多。
如果某物種的基因組全序列已經破譯,并不代表該物種的蛋白質組也已破譯。 具體分析某個基因的蛋白質產物要綜合基因組水平、轉錄水平和翻譯水平的修飾及調控來確定。
研究內容
主要有兩方面,一是結構蛋白質組學,二是功能蛋白質組學。其研究前沿大致分為三個方面:
① 針對有關基因組或轉錄組數據庫的生物體或組織細胞,建立其蛋白質組或亞蛋白質組及其蛋白質組連鎖群,即組成性蛋白質組學。
② 以重要生命過程或人類重大疾病為對象,進行重要生理病理體系或過程的局部蛋白質組或比較蛋白質組學。
③ 通過多種先進技術研究蛋白質之間的相互作用,繪制某個體系的蛋白,即相互作用蛋白質組學,又稱為“細胞圖譜”蛋白質組學。
此外,隨著蛋白質組學研究的深入,又出現了一些新的研究方向,如亞細胞蛋白質組學、定量蛋白質組學等。蛋白質組學是系統生物學的重要研究方法.
技術原理
雙向凝膠電泳技術(2-DE)
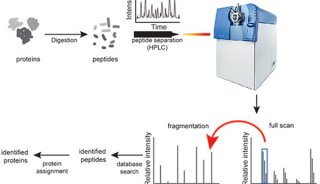
雙向凝膠電泳技術與質譜技術是目前應用最為廣泛的研究蛋白質組學的方法。雙向凝膠電泳技術利用蛋白質的等電點和分子量差別將各種蛋白質區分開來。雖然二維凝膠電泳難以辨別低豐度蛋白,對操作要求也較高,但其通量高、分辨率和重復性好以及可與質譜聯用的特點,使其成為目前最流行、可靠的蛋白質組研究手段。雙向凝膠電泳技術及質譜基礎的蛋白質組學研究程序為樣品制備→等電聚焦→聚丙烯酰胺凝膠電泳→凝膠染色→挖取感興趣的蛋白質點→膠內酶切→質譜分析確定肽指紋圖譜或部分氨基酸序列→利用數據庫確定蛋白。蛋白質組研究要求有高分辨率的蛋白質分離及準確、靈敏的質譜鑒定技術。凝膠電泳中蛋白質的著色不僅影響蛋白質分離的分辨率,同時也影響后續的質譜鑒定。蛋白質的染色可分為有機試劑染色、銀染、熒光染色及同位素顯色四類。
Unlu 等提出了一種熒光差異顯示雙向電泳(F-2D-DIGE)的定量蛋白質組學分析方法。差異凝膠電泳(DIGE)是對2-DE 在技術上的改進,結合了多重熒光分析的方法,在同一塊膠上共同分離多個分別由不同熒光標記的樣品,并第一次引入了內標的概念。兩種樣品中的蛋白質采用不同的熒光標記后混合,進行2-DE,用來檢測蛋白質在兩種樣品中表達情況,極大地提高了結果的準確性、可靠性和可重復性。在DIGE技術中,每個蛋白點都有它自己的內標,并且軟件可全自動根據每個蛋白點的內標對其表達量進行校準,保證所檢測到的蛋白豐度變化是真實的。DIGE 技術已經在各種樣品中得到應用。
高效液相色譜技術(HPLC)
盡管二維凝膠電泳(2-DE)是常用的對全蛋白組的分析方法,但其存在分離能力有限、存在歧視效應、操作程序復雜等缺陷。對于分析動態范圍大、低豐度以及疏水性蛋白質的研究往往很難得到滿意的結果。Chong 等使用HPLC/ 質譜比較分析惡性腫瘤前和癌癥兩種蛋白質差異表達。利用HPLC 分離蛋白質,并用MALDI-TOF-MS鑒定收集的組分,從而在兩種細胞中的差異表達中對蛋白質進行定量分析。多維液相色譜作為一種新型分離技術,不存在相對分子質量和等電點的限制,通過不同模式的組合,消除了二維凝膠電泳的歧視效應,具有峰容量高、便于自動化等特點。二維離子交換-反相色譜(2D-IEC-RPLC)是蛋白質組學研究中最常用的多維液相色譜分離系統。
質譜技術
最早是MALDI-TOF,MALDI基質輔助激光解吸離子化技術于2002 年由諾貝爾化學獎得主田中發明,剛剛產生便引起學術界的高度重視。MALDI和TOF(飛行時間質譜)搭配是一個理想的快速鑒定技術。最早,SELDI 技術是蛋白質組學研究中比較理想的技術平臺,其全稱是表面增強激光解吸電離飛行時間質譜技術(SELDI-tof)。其方法主要如下:通常情況下將樣品經過簡單的預處理后直接滴加到表面經過特殊修飾的芯片上,既可比較兩個樣品之間的差異蛋白,也可獲得樣品的蛋白質總覽。因此,在應用方面具有顯著優勢。SELDI 技術分析的樣品不需用液相色譜或氣相色譜預先純化,因此可用于分析復雜的生物樣品。SELDI 技術可以分析疏水性蛋白質,PI 過高或過低的蛋白質以及低分子質量的蛋白質( < 25 000) ,還可以發現在未經處理的樣品中許多被掩蓋的低濃度蛋白質,增加發現生物標志物的機會。SELDI 技術只需少量樣品,在較短時間內就可以得到結果,且試驗重復性好,適合臨床診斷及大規模篩選與疾病相關的生物標志物,特別是它可直接檢測不經處理的尿液、血液、腦脊液、關節腔滑液、支氣管洗出液、細胞裂解液和各種分泌物等, 從而可檢測到樣品中目標蛋白質的分子量、PI、糖基化位點、磷酸化位點等參數。
后來,人們發現SELDI-TOF的數據并不是很理想,由于蛋白質組學的復雜性,SELDI-TOF號稱的種種“發現腫瘤Pattern”等被證明是不可靠的。SELDI-TOF當時的過度宣傳,和馬上被證實的不可靠,被人們拋入了谷底,打入了冷宮。目前,人們認為如果采用MALDI技術,至少要接串聯質譜。
在初期的高通量鑒定時代,第一個獲勝的技術是MALDI-TOF/TOF,它和2D?PAGE連接,實現了生物學家更容易理解的,跑膠-分離提取-質譜鑒定的流程。
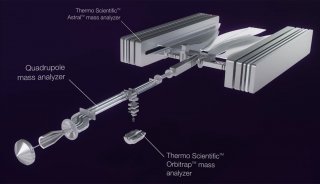
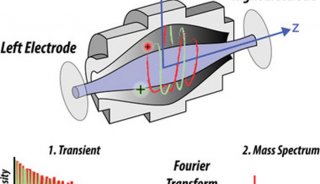
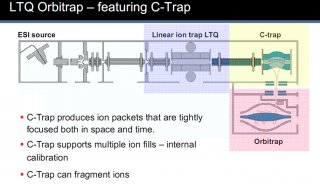
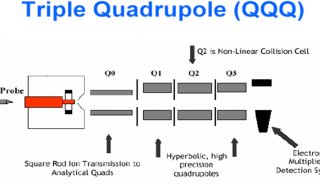
與此競爭的技術是2D?HPLC-MS(這個MS可以是:LCQ、LTQ、Q-TOF、Orbitrap),這些也都是串聯質譜技術。后來2D?HPLC-MS技術逐步戰勝了MALDI-TOF/TOF技術,因為跑膠,不能在線,也很難重現。當2D?HPLC技術克服了重重難關后,越來越多的人放棄了MALDI-TOF/TOF技術。
在高通量鑒定時代之后,進入了更多的Biomarker發現,定量,磷酸化等翻譯后修飾鑒定,蛋白-蛋白相互作用等時代。這時的精細鑒定,更需要LC-MS技術,MALDI-TOF/TOF技術就更被人們暫時拋到腦后。直到最近,人們步入到質譜成像階段,越來越多的醫學工作者更加容易信任成像技術,而MALDI成像相對于基于ESI的質譜,更容易實現穩定可靠的成像。
同位素標記親和標簽(ICAT)技術
同位素親和標簽技術是一種用于蛋白質分離分析技術,此技術是蛋白質組研究技術中的核心技術之一。該技術用具有不同質量的同位素親和標簽( ICATs) 標記處于不同狀態下的細胞中的半胱氨酸,利用串聯質譜技術,對混合的樣品進行質譜分析。來自兩個樣品中的同一類蛋白質會形成易于辨識比較的兩個不同的峰形,能非常準確的比較出兩份樣品蛋白質表達水平的不同。ICAT 的好處在于它可以對混合樣品直接測試;能夠快速定性和定量鑒定低豐度蛋白質,尤其是膜蛋白等疏水性蛋白等;還可以快速找出重要功能蛋白質。
由于采用了一種全新的ICAT試劑,同時結合了液相色譜和串聯質譜,因此不但明顯彌補了雙向電泳技術的不足,同時還使高通量、自動化蛋白質組分析更趨簡單、準確和快速,代表著蛋白質組分析技術的主要發展方向。針對磷酸化蛋白分析以及與固相技術相結合ICAT技術本身又取得了許多有意義的進展,已形成ICA T 系列技術。
生物信息學技術
生物信息學在生命科學研究中起著越來越重要的作用。利用生物信息學對蛋白質組的各種數據進行處理和分析,也是蛋白質組研究的重要內容。生物信息學是蛋白質組學研究中不可缺少的一部分。生物信息學的發展,已不僅是單純的對基因組、蛋白質組數據的分析,而且可以對已知的或新的基因產物進行全面分析。在蛋白質組數據庫中儲存了有機體、組織或細胞所表達的全部蛋白質信息,通過用鼠標點擊雙向凝膠電泳圖譜上的蛋白質點就可獲得.
鑒定方法
如蛋白質鑒定結果、蛋白質的亞細胞定位、蛋白質在不同條件下的表達水平等信息。目前應用最普遍的數據庫是NRDB和dbEST 數據庫。NRDB由SWISS2PROT 和GENPETP 等幾個數據庫組成,dbEST是由美國國家生物技術信息中心(NCBI)和歐洲生物信息學研究所(EBI)共同編輯的核酸數據庫;計算機分析軟件主要有蛋白質雙向電泳圖譜分析軟件、蛋白質鑒定軟件、蛋白質結構和功能預測軟件等。
研究進展
2014年5月28日,英國新一期《自然》雜志公布兩組科研人員分別繪制的人類蛋白質組草圖。這一成果有助于了解各個組織中存在何種蛋白質,這些蛋白質與哪些基因表達有關等,從而進一步揭開人體的奧秘。?
上世紀90年代,人類基因組計劃開始成形時,有科學家提出了破譯人類蛋白質組的想法。其目標是將人體所有蛋白質歸類并描繪出它們的特性、在細胞中所處的位置以及蛋白質之間的相互作用。但人類蛋白質組的規模和復雜性使此類研究困難重重。?
研究人員借助計算機對這些蛋白質片段與基因組進行了大量比對工作,并據此列出一個“清單”,描繪出哪些組織中的哪些基因表達與蛋白質的形成有關。在另一項研究中,美國約翰斯·霍普金斯大學研究人員與印度等國同行也采用質譜分析法繪制出一張蛋白質組草圖。?

-
企業風采
-
企業風采

-
會議會展