分享至
分享至



Nature | 炎癥性腸病腸道微生物群的多重組學研究
炎癥性腸病(Inflammatory bowel diseases,IBD)影響著全世界超過350萬人,其發病率逐年增加。IBD最常見的形式是克羅恩病(CD)和潰瘍性結腸炎(UC),在臨床、免疫學、分子、遺傳和微生物水平上具有異質性,其特征是CD或UC的炎癥減弱、慢性復發、虛弱、胃腸道或者結腸炎癥緩解。它是由宿主、微生物和環境因素之間復雜相互作用的結果。IBD患者腸道微生物群的常見變化是兼性厭氧菌(如大腸桿菌)增加,產生短鏈脂肪酸(SCFAs)的專性厭氧菌減少。哈佛大學Curtis Huttenhower研究團隊建立Inflammatory Bowel Disease Multi’omics Database(IBDMDB),作為整合人類微生物組項目的一部分,以強化對IBD腸道微生物組病因學的系統理解,相關研究成果發表于《Nature》。

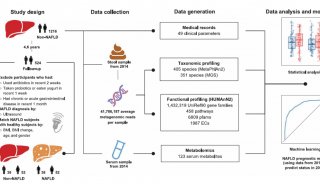
本研究從五個學術醫療中心(三個兒科隊列和兩個成人隊列,Fig.1a)招募了132名參與者并隨訪1年,根據初始內窺鏡和組織病理學結果未診斷為IBD的個體被歸為非IBD對照組(Non-IBD),共收集24個時間點的651例活檢,529例血樣和1785例糞便樣本進行宏基因組(MGX),元轉錄組(MTX),蛋白質組(MPX),代謝組(MBX)和病毒體(VX)檢測(Fig.1b),得到305個樣本的全部測量分析數據結果及791個MGX- MTX結果(Fig.1c)。
IBD腸道微生物群的多組學變化
主坐標分析顯示第二主坐標軸更能區分IBD,特別是CD,且大多數變異是由擬桿菌門和厚壁菌門之間的權衡引起的,IBD個體的α多樣性低于Non-IBD(圖1d)。大多數測試數據在受試者間、受試者內橫斷面和縱向上存在相關性,從MGX,MTX和MPX中獲得的功能譜是聯系最緊密的(圖1e),IBD和Non-IBD患者間的橫斷面差異在代謝組中最為明顯(圖1f, 2a)。這可能是由于營養吸收不良、腸內水分或血液含量增加以及IBD個體腸內轉運時間縮短造成的。IBD患者的多不飽和脂肪酸(腎上腺素和花生四烯酸)更為豐富,而IBD患者腸道中泛酸鹽和煙酸鹽(分別為維生素B5和B3)缺乏,而煙酸鹽在腸道中具有抗炎和抗凋亡的功能,此外,IBD患者血清很少存在B族維生素缺乏癥狀。
糞便鈣衛蛋白和Harvey Bradshaw指數(HBI)是CD疾病嚴重程度的兩個指標,UC中的簡單臨床結腸炎活動指數(SCCAI)與糞便鈣衛蛋白水平相關性較弱(圖2b)。微生物失調評分通常作為疾病活動的衡量指標,通過測量血清ASCA(抗釀酒酵母抗體)、ANCA(抗中性粒細胞胞質抗體)、OmpC(外膜蛋白C)和CBir1(抗鞭毛蛋白)抗體滴度的高低來評判。在生態失調期間的分類學擾動同樣反映了在IBD橫斷面觀察到的現象,如CD中的專性厭氧菌(Faecalibacterium prausnitzii,?Roseburia hominis)的消耗以及大腸桿菌等兼性厭氧菌的富集(圖2f)。IBD中比較突出的兩種瘤胃球菌Ruminococcus torques,?Ruminococcus gnavus在失調的CD和UC中的含量也存在顯著差異。

在代謝組中,SCFAs通常在生態失調組中減少,丁酸鹽的減少與丁酸鹽產生菌如F. prausnitzii和R. hominis的消耗相一致(圖2f)。CD生態失調組中初級膽酸鹽、甘氨酸/牛磺酸結合型膽汁酸、甘氨鵝脫氧膽酸出現富集;次級膽汁酸石膽酸鹽和脫氧膽酸鹽的減少,表明產生次級膽汁酸的細菌在IBD生態失調中被耗盡或者通過結腸的轉運時間太短,這些化合物不能代謝。此外許多酰基肉堿在生態失調中顯著富集,而基礎代謝物的水平通常會降低(圖2f)。然而花生四烯酰肉堿(C20:4肉毒堿)減少,游離花生四烯酸(參與炎癥的前列腺素的前體)增加(圖2a)。與膽汁酸一樣,肉毒堿也是微生物修飾的化合物,如左旋肉堿往往具有抗炎作用,而脂肪酸結合的肉毒堿對腸道炎癥不起作用。在失調的IBD個體中,許多其他代謝物也顯著改變(檢測到的548種代謝物中有117種具有顯著差異),代謝產物池的大規模失調與宿主和微生物的分類學和分子特征相一致(圖2f),微生物生態失調期間的顯著代謝組學差異與疾病期間預期的變化一致,進一步表明生態失調與IBD相關。

腸內微生物群穩定性下降
IBD中微生物群的時間變化更頻繁,通過宏基因組學、宏轉錄組學和代謝組學譜分析,每個受試者的微生物組隨時間偏離基線(Fia.3a)。作者通過尋找連續時間點之間微生物群的‘shifts’(Bray Curtis差異,用于描述不同個體的差異而非個體內部差異)來表征大規模的時間差異(圖3a)。首先,僅考慮宏基因組分類學特征,發現166個‘shifts’(Non-IBD, 39; UC, 44; CD, 83),CD或UC患者的轉換率略高于Non-IBD(2.09,1.83,1.79/年)。相對豐度變化最大的物種差異顯著(圖3b),Non-IBD個體的移位主要發生在豐度高的普氏菌屬(Prevotella copri)個體中,其在數周至數月豐度反復波動,IBD患者的相對豐度更穩定(圖3c)。IBD的分類學變化反映了早期觀察到的專性厭氧菌的相對減少和兼性厭氧菌的過度生長(圖3b),并與進入和退出生態失調相對應,如大腸桿菌促成了IBD的大量轉移。

代謝組學的轉換率大約是宏基因組的一半(Non-IBD, 1.05; UC, 0.99; CD, 1.36),檢測了相同受試者相鄰樣本之間代謝物譜的差異,并通過診斷發現了顯著的分離(圖3d)。尿膽素在Non-IBD的個體中具有最大差異,CD患者中差異大的尿酸鹽,及m/z 152.0354且RT為4.16 min的feature(可能吡啶醛的甲酸加合物)很大程度上與UC有關。其中甲咪唑乙酸和尿酸鹽是最主要的轉移因子(圖3 e),當然未知代謝物是導致差異的主要原因,需要進一步的開展代謝物注釋來確定未知代謝物在IBD中的重要性,如HILp_QI22918, m/z值為648.43067, RT為5.03 min的feature在IBD患者中貢獻最為顯著。
微生物相關的宿主因子
作者將基線時結腸鏡下的腸道活檢結果納入IBD中微生物組的分析,對每個活檢取樣部位進行單獨的微生物組和表型關聯分析。相比Non-IBD,在CD回腸炎癥部位鑒定了305個基因,其中IL-17信號富集最強;CD和UC直腸炎癥部位鑒定了920個存在顯著差異表達的基因(DEGs)(Fig.4a),包括可直接影響共生微生物的基因如抗菌CXCL6(細胞膜破壞因子),SAA2(抑制革蘭氏陰性菌的生長)及間接微生物調節劑如DUOX2(雙氧酶,產生活性氧),LCN2(通過隔離誘導微生物鐵饑餓)(圖4b)。為了確定微生物組中與這些變化最相關的成分,相同的樣本進行16S測序,分別在回腸和直腸中鑒定出31和106對顯著的OTU對,Spearman相關性分析顯示DUOX2及其成熟因子DUOXA2均與回腸中瘤胃球菌科UCG 005 (OTU 89)的豐度呈負相關。幾種趨化因子基因的表達顯示抗菌劑(CXCL6, ?CCL20)與直腸中的短桿菌 (OTU 120)、鏈球菌(OTU 37) 和Eikenella (OTU 39) 的相對豐度呈負相關,表明這些物種對上述趨化因子的活性最敏感。
動態的多元微生物相互作用
接下來作者采用宏基因組物種、物種水平轉錄率、酶基因家族(MGX, MTX, MPX)功能譜、代謝物、宿主轉錄(直腸和回腸),血清學和糞便鈣衛蛋白的數據構建關聯網絡來搜索可能構成IBD疾病活動的宿主和微生物分子相互作用。為了確定與炎癥和疾病狀態密切相關的共變異,首先使用混合效應模型對每種測量類型進行殘差分析確定差異豐度,得到53161條邊和2916個節點,再構建過濾子網對每個測量類型的前300條邊(按P值)進行可視化,其中至少一個連接節點與生態失調相關(圖4c)。具有至少20個連接的節點作為該網絡的中心(Hubs),認為在生態失調中差異豐富。F. prausnitzii是關聯最強的分類學特征,還有未被分類的Subdoligranulum,均與膽固醇和肌苷的豐度相關,許多ECs的表達在失調中被下調。
酰基肉堿和膽汁酸在網絡中占據突出地位。大腸桿菌占上調ECs的大部分,Roseburia(與Subdoligranulum一起)參與IBD中肉毒堿和膽汁酸的失調。酰基肉堿與許多物種生態失調相關如人葡萄球菌(9種酰基肉堿),肺炎克雷伯氏菌(3種)和副流感嗜血桿菌(3種),C. bolteae(3種)的表達。C8肉毒堿是網絡中特別值得關注的生物化學中心,另一種在失調CD中顯著增加的酰基肉堿,和膽酸鹽、鵝脫氧膽酸鹽、牛磺酸脫氧膽酸鹽共占了107個邊緣(6%;圖4c)。其他重要的代謝物關聯包括幾個長鏈脂質中樞和SCFA丙酸鹽; 抗OmpC抗體與眾多ECs的宏基因組豐度密切相關;鈣衛蛋白與生態失調中含量差異不顯著的代謝物及ECs的宏基因組豐度弱相關。在這個重要的子網絡中出現了三個宿主基因(GIP,NXPE4,ANXA10)的回腸表達。RNA聚合酶的表達也是網絡中的一個突出節點,在生態失調中被上調。本研究將IBD中多種類型的微生物群破壞與許多分子特征聯系起來,這些分子特征將成為后續研究IBD和胃腸道炎癥機制的潛在目標。

小結
本研究開發的IBDMDB是對IBD動力學中涉及的腸道微生物組的多個分子特征的首批綜合研究之一。作為整合人類微生物組項目(HMP2或iHMP)的一部分,作者對132名受試者進行了為期一年的跟蹤研究,以獲得疾病期間宿主和微生物活動的縱向綜合分子譜(各24個時間點,共2965例糞便、活檢和血液標本)。在腸道炎癥疾病活動期間腸道微生物功能失調,證明了兼性厭氧菌的特征性增加是以犧牲專性厭氧菌為代價,微生物轉錄(例如梭狀芽孢桿菌)、代謝產物池(酰基肉堿、膽汁酸和短鏈脂肪酸)和宿主血清抗體水平的分子紊亂。在確定代謝物池的變化是宿主驅動的還是微生物驅動的之后,酰基肉堿失調也可能成為IBD治療的潛在新靶點,疾病活動期以時間變異性的增加為特征,具有特征性的分類、功能和生化變化。最后,綜合分析確定了導致這種失調的微生物、生化和宿主因素。本文提供了迄今為止最全面的炎癥性腸病宿主和微生物的測量數據,最重要的是將這些分子結果帶回臨床,以更好地預測IBD進展和結果的生物標志物,并作為一組新的宿主微生物相互作用靶點,從而開發出治療IBD的方法。
參考文獻
Jason L P, Curtis H, et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 569, 655–662 (2019). https://doi.org/10.1038/s41586-019-1237-9.
微信公眾號:麥特繪譜
Tel:400-867-2686
Email : marketing@metaboprofile.com
Web:?www.metaboprofile.com


-
儀器推薦

-
儀器推薦
-
儀器推薦
 詢底價 Tel:400-6699-117 轉 6306
詢底價 Tel:400-6699-117 轉 6306 -
儀器推薦
-
廠商文章

-
廠商文章

-
廠商文章

-
廠商文章

-
廠商文章

-
廠商文章

-
廠商文章

-
廠商文章

-
廠商文章
-
廠商文章

-
廠商文章

-
廠商文章

-
廠商文章

-
廠商文章