生成式AI加速抗病毒藥物開發,提高藥物發現潛力
新興藥物靶蛋白的抑制劑發現具有挑戰性,特別是當靶結構或活性分子未知時。
IBM 研究院、牛津大學和 Diamond Light Source 公司的合作團隊,通過實驗驗證了深度生成框架的廣泛實用性,他們的框架在蛋白質序列、小分子及其相互相互作用上進行了大規模訓練,不偏向任何特定目標。
研究人員在生成基礎模型上進行了蛋白質序列條件采樣,為兩個不同的靶點設計小分子抑制劑:刺突蛋白受體結合域(RBD)和 SARS-CoV-2 的主要蛋白酶。
盡管在模型推斷過程中僅使用靶序列信息,但在每個靶合成的四個候選物中,有兩個在體外觀察到微摩爾水平的抑制。最有效的刺突 RBD 抑制劑在活病毒中和試驗中表現出針對多種變體的活性。這表明,即使在缺乏目標結構或結合物信息的情況下,用于加速抑制劑發現的單一的、可廣泛部署的生成基礎模型也是有效且高效的。
該研究以「Accelerating drug target inhibitor discovery with a deep generative foundation model」為題,于 2023 年 6 月 21 日發布在《Science Advances》。

從頭分子設計具有挑戰性
從頭分子設計,即提出具有所需特性的先前未識別的化合物,是藥物發現和材料工程應用中的一個具有挑戰性的問題。
例如,尋找那些作為進一步設計候選藥物的化學起點的抑制劑化合物,通常涉及采用對含有標準化合物或較小化學片段的文庫進行高通量篩選,但這類方法的成功率在 0.5% 到 1% 之間,具體取決于篩選的文庫大小(通常約為 10^4 個條目)和目標特征。
成功率低的部分原因是搜索空間巨大,目前估計涵蓋 10^33 到 10^80 個可行分子,其中通常只有一小部分具有所尋求的特征;因此通過實驗來依次篩選是不可行的。
除了需要數千次篩選實驗之外,文庫的初始選擇通常還需要與已報道的配體結合的目標蛋白的詳細結構信息,而這些信息通常不容易獲得。最后,由于基礎設施、化合物和試劑的成本,導致抑制劑發現可能非常昂貴。
因此,迫切需要一種更有效的方法,以便能夠從廣闊的化學空間(包括尚未合成的分子)中蒸餾出以前未識別的和有前途的分子。這種方法將能夠對一小部分候選藥物進行實驗驗證,從而以更少的時間和成本提高抑制劑的發現率。
DL可應對挑戰但也有局限性
基于深度學習的生成模型有可能以「無規則」的方式發現具有所需功能的先前未識別的分子,因為他們的目標是首先學習已知化學物質的密集、連續表示(以下稱為潛在向量),然后修改潛在向量以解碼為看不見的分子。因此,此類模型提供了進入以前未探索的化學空間的機會,不受人類有意識偏見的限制。
然而,對于目標特異性藥物樣抑制劑設計的任務,必須使用「反向分子設計」方法,其中通過學習的化學表示的導航是由分子屬性屬性引導的,例如目標抑制活性和藥物相似性。在針對先前未識別的靶標設計抑制劑的情況下,需要足夠量的示范分子,而這可能是無法獲得的,并且需要昂貴且耗時的篩選實驗才能獲得。
由于大多數現有的深度生成框架仍然依賴于從特定目標的結合劑化合物庫中學習,因此它們限制了對已知和整體分子的固定庫之外的探索,同時阻止了機器學習框架對先前未識別的目標的泛化。
因此,雖然一些使用深度生成模型進行目標特異性抑制劑設計的研究已經過實驗驗證,但尚未有報道稱這些模型能夠在不同的蛋白質靶點上發現經過驗證的抑制劑,而無需獲得詳細的靶點特異性先前結合數據(例如靶點結合劑分子)。
一種新的深度生成模型
IBM 研究院、牛津大學和 Diamond Light Source 公司的聯合團隊,展示了基于深層生成基礎模型的單一、統一的抑制劑設計框架在不同靶蛋白上的現實應用性。生成框架只需要更容易獲得的目標序列信息來指導設計。此外,該工作考慮了(i)設計命中的脫靶結合,以考慮潛在的下游不利影響,(ii)即使在未知結合物的情況下也能識別命中,以及(iii)優先考慮易于合成的化合物。
「開發和驗證這些方法需要時間,但現在我們有了工作流程,可以更快地生成結果。」 該研究的共同高級作者、IBM 研究院研究員 Payel Das 說,「當下一種病毒出現時,生成人工智能可能在尋找新療法中發揮關鍵作用。」
論文的聯合資深作者 Martin Walsh 表示:「生成與感興趣的藥物靶標具有高親和力結合的初始化合物,可以加速基于結構的藥物發現流程,并支持我們為未來的流行病做好更好準備的努力。」
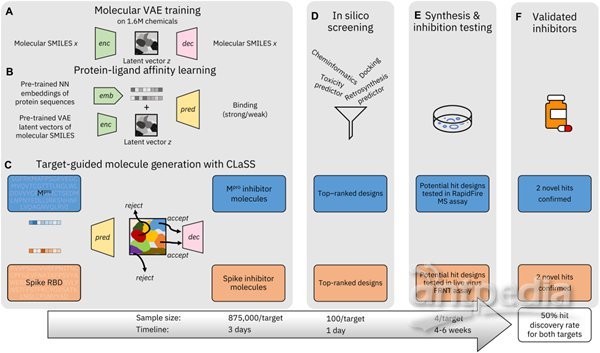
由 CogMol 驅動的抑制劑發現工作流程概述
研究人員使用 CogMol(一種深度生成模型)針對兩個重要且獨特的 SARS-CoV-2 靶標提出了先前未識別的且化學上可行的抑制劑設計:刺突 (S) 蛋白和主蛋白酶 (M^pro) 蛋白的受體結合域 (RBD)。
深層生成框架建立在化學分子、蛋白質序列和蛋白質-配體結合數據的大規模數據之上,作為目標感知抑制劑分子設計的生成基礎模型,無需對特定目標數據進行任何進一步微調,并且可以外推到原始訓練數據中不存在的目標序列。因此,CogMol 框架的這種廣泛通用性將其置于新興的「基礎模型」類別中,這些模型是在大量未標記數據上進行預訓練的,并且可以通過最少的微調用于不同的下游任務。
由 CogMol 設計的一組先前未識別的針對 SARS-CoV-2 蛋白的分子,于 2020 年 4 月在知識共享許可下在 IBM COVID-19 Molecule Explorer 平臺上共享。在這里,研究人員通過合成和測試一些針對 S 蛋白和 M^pro 蛋白的 SARS-CoV-2 RBD 的優先設計的抑制活性,對 CogMol 深度生成框架的廣泛實用性和就緒性進行了首次實驗驗證。
該團隊對類先導化學物質庫進行虛擬篩選,進一步證明了 CogMol 框架中使用的結合親和力預測模型的適用性,并通過晶體學分析成功鑒定出三種化合物,證實其結合在M^pro的活性位點上,其中一種化合物表現出微摩爾抑制作用。
該研究首次提供了單一生成機器智能框架的經過驗證的演示,該框架可以在設計過程中僅使用蛋白質序列信息,以高成功率為不同的蛋白質藥物靶點提出先前未識別的有前途的抑制劑。
所設計的刺突抑制劑針對所關注的 SARS-CoV-2 變體表現出廣譜抗病毒活性,進一步確立了這種深層生成框架加速和自動化命中發現周期的潛力,該過程已知產量低、損耗率高,但也增進了研究人員對較少探索的藥物靶點的科學認識。
「我們使用生成基礎模型為加速抗病毒藥物的開發創造了有效的起點,而該模型對其蛋白質靶標知之甚少。」 該研究的共同高級作者、IBM 研究院研究員、牛津大學教授 Jason Crain 說,「我希望這些方法將使我們能夠在未來更快、更便宜地制造抗病毒藥物和其他急需的化合物。」